Calculations
Here the calculations of the aiida-kkr plugin are presented. It is assumed that the user already has basic knowledge of python, aiida (e.g. database structure, verdi commands, structure nodes) and KKR (e.g. LMAX cutoff, energy contour integration). Also aiida-kkr should be installed as well as the Voronoi, KKR and KKRimp codes should already be configured.
In practice, the use of the workflows is more convenient but here the most basic calculations which are used underneath in the workflows are introduced step by step.
In the following the calculation plugins provided by aiida-kkr are introduced at the example of bulk Cu.
Note
If you follow the steps described here please make sure that your python script contains:
from aiida import load_profile
load_profile()
To ensure that the aiida database is properly integrated.
Voronoi starting potential generator
The Voronoi code creates starting potentials for a KKR calculation and sets up the atom-centered division of space into voronoi cells. Also corresponding shape functions are created, which are needed for full-potential corrections.
The voronoi plugin is called kkr.voro and it has the following input and output nodes:
- Three input nodes:
parametersKKR parameter set for Voronoi calculation (Dict)structurestructure data node node describing the crystal lattice (StructureData)codeVoronoi code node (code)
- Three output nodes:
remote_folder(RemoteData)retrieved(FolderData)output_parameters(Dict)
- Additional optional input nodes that trigger special behavior of a Voronoi calculation are:
parent_KKR(RemoteData of a KKR Calculation)potential_overwrite(SingleFileData)
Now the basic usage of the voronoi plugin is demonstrated at the example of Cu bulk for which first the aiida structure node and the parameter node containing KKR specific parameters (LMAX cutoff etc.) are created before a voronoi calculation is set up and submitted.
Input structure node
First we create an aiida structure:
# get aiida StructureData class:
from aiida.plugins import DataFactory
StructureData = DataFactory('structure')
Then we create the aiida StructureData node (here for bulk Cu):
alat = 3.61 # lattice constant in Angstroem
bravais = [[0.5*alat, 0.5*alat, 0], [0.5*alat, 0, 0.5*alat], [0, 0.5*alat, 0.5*alat]] # Bravais matrix in Ang. units
# now create StructureData instance and set Bravais matrix and atom in unit cell
Cu = StructureData(cell=bravais)
Cu.append_atom(position=[0,0,0], symbols='Cu')
Input parameter node
Next we create an empty set of KKR parameters (LMAX cutoff etc. ) for voronoi code:
# load kkrparms class which is a useful tool to create the set of input parameters for KKR-family of calculations
from masci_tools.io.kkr_params import kkrparams
params = kkrparams(params_type='voronoi')
Note
we can find out which parameters are mandatory to be set using
missing_params = params.get_missing_keys(use_aiida=True)
and set at least the mandatory parameters:
params.set_multiple_values(LMAX=2, NSPIN=1, RCLUSTZ=2.3)
finally create an aiida Dict node and fill with the dictionary of parameters:
Dict = DataFactory('dict') # use DataFactory to get ParamerterData class
ParaNode = Dict(dict=params.get_dict())
Submit calculation
Now we get the voronoi code:
from aiida.orm import Code # load aiida 'Code' class
codename = 'voronoi@localhost'
code = Code.get_from_string(codename)
Note
Make sure that the voronoi code is installed:
verdi code list should give you a list of installed codes where codename should be in.
and create new process builder for a VoronoiCalculation:
builder = code.get_builder()
Note
This will already set builder.code to the voronoi code which we loaded above.
and set resources that will be used (here serial job) in the options dict of the metadata:
builder.metadata.options = {'resources': {'num_machines':1, 'tot_num_mpiprocs':1} }
Note
If you use a computer without a default queue you need to set the name of the queue as well:
builder.metadata.options['queue_name'] = 'th1')
then set structure and input parameter:
builder.structure = Cu
builder.parameters = ParaNode
Note
Additionally you could set the parent_KKR and potential_overwrite input nodes which trigger special run modes of the voronoi code that are discussed below.
Now we are ready to submit the calculation:
from aiida.engine import submit
voro_calc = submit(builder)
Note
check calculation state (or use verdi calculation list -a -p1) using
voro_calc.process_state
Voronoi calculation with the parent_KKR input node
To come …
Voronoi calculation with the potential_overwrite input node
To come …
KKR calculation for bulk and interfaces
A KKR calculation is provided by the kkr.kkr plugin, which has the following
input and output nodes.
- Three input nodes:
parametersKKR parameter fitting the requirements for a KKR calculation (Dict)parent_folderparent calulation remote folder node (RemoteFolder)codeKKR code node (code)
- Three output nodes:
remote_folder(RemoteData)retrieved(FolderData)output_parameters(Dict)
Note
The parent calculation can be one of the following:
Voronoi calculation, initial calculation starting from structure
previous KKR calculation, e.g. preconverged calculation
The necessary structure information is always extracted from the voronoi parent calculation. In case of a continued calculation the voronoi parent is recuresively searched for.
- Special features exist where a fourth input node is persent and which triggers special behavior of the KKR calculation:
impurity_infoNode specifying the impurity cluster (Dict)kpointsNode specifying the kpoints for which the bandstructure is supposed to be calculated (KpointsData)
The different possible modes to run a kkr calculation (start from Voronoi calculation, continue from previous KKR calculation, host Greenfunction writeout feature) are demonstrated in the following.
Start KKR calculation from voronoi parent
Reuse settings from voronoi calculation:
voronoi_calc_folder = voro_calc.out.remote_folder
voro_params = voro_calc.inputs.parameters
Now we update the KKR parameter set to meet the requirements for a KKR calculation (slightly different than voronoi calculation). Thus, we create a new set of parameters for a KKR calculation and fill the already set values from the previous voronoin calculation:
# new kkrparams instance for KKR calculation
params = kkrparams(params_type='kkr', **voro_params.get_dict())
# set the missing values
params.set_multiple_values(RMAX=7., GMAX=65.)
# choose 20 simple mixing iterations first to preconverge potential (here 5% simple mixing)
params.set_multiple_values(NSTEPS=20, IMIX=0, STRMIX=0.05)
# create aiida Dict node from the KKR parameters
ParaNode = Dict(dict=params.get_dict())
Note
You can find out which parameters are missing for the KKR calculation using params.get_missing_keys()
Now we can get the KKR code and create a new calculation instance and set the input nodes accordingly:
code = Code.get_from_string('KKRcode@localhost')
builder = code.get_builder()
# set input Parameter, parent calulation (previous voronoi calculation), computer resources
builder.parameters = ParaNode
builder.parent_folder = voronoi_calc_folder
builder.metadata.options = {'resources' :{'num_machines': 1, 'num_mpiprocs_per_machine':1}}
We can then run the KKR calculation:
kkr_calc = submit(builder)
Continue KKR calculation from KKR parent calculation
First we create a new KKR calculation instance to continue KKR ontop of a previous KKR calclation:
builder = code.get_builder()
Next we reuse the old KKR parameters and update scf settings (default is NSTEPS=1, IMIX=0):
params.set_multiple_values(NSTEPS=50, IMIX=5)
and create the aiida Dict node:
ParaNode = Dict(dict=params.get_dict())
Then we set the input nodes for calculation:
builder.parameters = ParaNode
kkr_calc_parent_folder = kkr_calc.outputs.remote_folder # parent remote folder of previous calculation
builder.parent_folder = kkr_calc_parent_folder
builder.metadata.options = {'resources': {'num_machines': 1, 'num_mpiprocs_per_machine':1}}
store input nodes and submit calculation:
kkr_calc_continued = submit(builder)
The finished calculation should have this output node that can be access within
python using kkr_calc_continued.outputs.output_parameters.get_dict(). An excerpt
of the ouput dictionary may look like this:
{u'alat_internal': 4.82381975,
u'alat_internal_unit': u'a_Bohr',
u'convergence_group': {
u'calculation_converged': True,
u'charge_neutrality': -1.1e-05,
u'nsteps_exhausted': False,
u'number_of_iterations': 47,
u'rms': 6.4012e-08,
...},
u'energy': -44965.5181266111,
u'energy_unit': u'eV',
u'fermi_energy': 0.6285993399,
u'fermi_energy_units': u'Ry',
u'nspin': 1,
u'number_of_atoms_in_unit_cell': 1,
u'parser_errors': [],
...
u'warnings_group': {u'number_of_warnings': 0, u'warnings_list': []}}
Special run modes: host GF writeout (for KKRimp)
Here we take the remote folder of the converged calculation to reuse settings and write out Green function and tmat of the crystalline host system:
kkr_converged_parent_folder = kkr_calc_continued.outputs.remote_folder
Now we extract the parameters of the kkr calculation and add the KKRFLEX run-option:
kkrcalc_converged = kkr_converged_parent_folder.get_incoming().first().node
kkr_params_dict = kkrcalc_converged.inputs.parameters.get_dict()
kkr_params_dict['RUNOPT'] = ['KKRFLEX']
The parameters dictionary is not passed to the aiida Dict node:
ParaNode = Dict(dict=kkr_params_dict)
Now we create a new KKR calculation and set input nodes:
code = kkrcalc_converged.inputs.code # take the same code as in the calculation before
builder= code.get_builder()
resources = kkrcalc_converged.attributes['resources']
builder.metadata.options = {'resources': resources}
builder.parameters = ParaNode
builder.parent_folder = kkr_converged_parent_folder
# prepare impurity_info node containing the information about the impurity cluster
imp_info = Dict(dict={'Rcut':4.0, 'ilayer_center': 0, 'Zimp':[79.]})
# set impurity info node to calculation
builder.impurity_info = imp_info
Note
The impurity_info node should be a Dict node and its dictionary should describe
the impurity cluster using the following parameters:
ilayer_center(int) layer index of position in the unit cell that describes the center of the impurity cluster
Rcut(float) cluster radius of impurity cluster in Ang. units
hcut(float, optional) height of a cylindrical cluster with radiusRcut, if not given spherical cluster is taken
cylinder_orient(list of 3 float values, optional)
Zimp(list of Nimp float entries) atomic charges of the substitutional impurities on positions defined byRimp_rel
Rimp_rel(list of Nimp [float, float, float] entries, optional, defaults to [0,0,0] for single impurity) cartesian positions of all Nimp impurities, relative to the center of cluster (i.e. position defined byilayer_center)
imp_cls(list of [float, float, float, int] entries, optional) full list of impurity cluster positions and layer indices (x, y, z, ilayer), overwrites auto generation usingRcutandhcutsettingsWarning
imp_clsfunctionality not implemented yet
Note
The retrieve_kkrflex node can be used to control wether or not the kkrflex_* files are copied back to the retrieved folder or only stay on the remote folder (saves space in the file repo).
The calculation can then be submitted:
# submit calculation
GF_host_calc = submit(builder)
Once the calculation has finished the retrieve folder should contain the kkrflex_* files needed for the impurity calculation.
Special run modes: bandstructure
Here we take the remote folder of the converged calculation and compute the bandstructure of the Cu bulk system. We reuse the DOS settings for the energy interval in which the bandstructure is computed from a previous calculation:
from aiida.orm import load_node
kkr_calc_converged = load_node(<-id-of-previous-calc>)
kkr_dos_calc = load_node(<-id-of-previous-DOS-calc>)
Now we need to generate the kpoints node for bandstructure calculation. This is
done using aiida’s get_explicit_kpoints_path function that extracts the kpoints
along high symmetry lines from a structure:
# first extract the structure node from the KKR parent calculation
from aiida_kkr.calculations.voro import VoronoiCalculation
struc, voro_parent = VoronoiCalculation.find_parent_structure(kkr_calc_converged.outputs.remote_folder)
# then create KpointsData node
from aiida.tools.data.array.kpoints import get_explicit_kpoints_path
kpts = get_explicit_kpoints_path(struc).get('explicit_kpoints')
Warning
Note that the get_explicit_kpoints_path function returns kpoints
for the primitive structure. In this example the input structure is already
the primitive cell however in general this may not always be the case.
Then we set the kpoints input node to a new KKR calculation and change some settings
of the input parameters accordingly (i.e. energy contour like in DOS run):
# create bandstructure calculation reusing old settings (including same computer and resources in this example)
kkrcode = kkr_calc_converged.inputs.code
builder = kkrcode.get_builder()
builder.kpoints = kpts # pass kpoints as input
builder.parent_folder = kkr_calc_converged.outputs.remote_folder
builder.metadata.options = {'resources': kkr_calc_converged.attributes['resources']}
# change parameters to qdos settings (E range and number of points)
from masci_tools.io.kkr_params import kkrparams
qdos_params = kkrparams(**kkr_calc_converged.inputs.parameters.get_dict()) # reuse old settings
# reuse the same emin/emax settings as in DOS run (extracted from input parameter node)
qdos_params.set_multiple_values(EMIN=host_dos_calc.inputs.parameters.get_dict().get('EMIN'),
EMAX=host_dos_calc.inputs.parameters.get_dict().get('EMAX'),
NPT2=100)
builder.parameters = Dict(dict=qdos_params.get_dict())
The calculation is then ready to be submitted:
# submit calculation
kkrcalc = submit(builder)
The result of the calculation will then contain the qdos.aa.s.dat files in the
retrieved node, where aa is the atom index and s the spin index of all atoms
in the unit cell. The resulting bandstructure (for the Cu bulk test system considered here)
should look like this (see here for the plotting script):
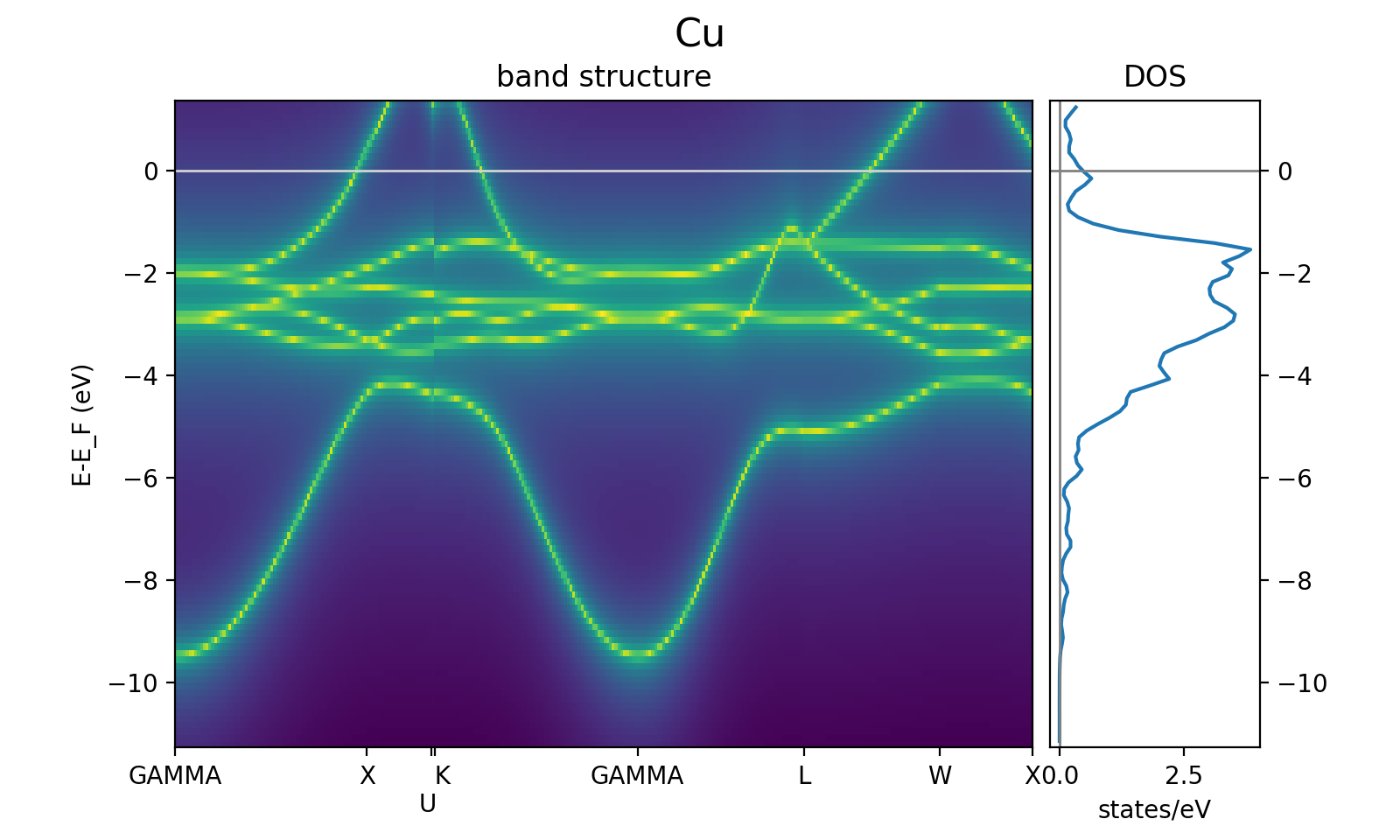
Special run modes: Jij extraction
The extraction of exchange coupling parameters is triggered with the XCPL
run option and needs at lest the JIJRAD paramter to be set.
Here we take the remote folder of the converged calculation and compute the exchange
parameters:
from aiida.orm import load_node
kkr_calc_converged = load_node(<-id-of-previous-calc>)
Then we set the XCLP run option and the JIJRAD parameter (the JIJRADXY,
JIJSITEI and JIJSITEJ parameters are not mandatory and are ommitted in this
example) in the input node to a new KKR calculation:
# create bandstructure calculation reusing old settings (including same computer and resources in this example)
kkrcode = kkr_calc_converged.inputs.code
builder = kkrcode.get_builder()
builder.parent_folder = kkr_calc_converged.outputs.remote_folder
builder.metadata.options = {'resources': kkr_calc_converged.attributes['resources']}
# change parameters to Jij settings ('XCPL' runopt and JIJRAD parameter)
from aiida_kkr.tools.kkr_params import kkrparams
Jij_params = kkrparams(**kkr_calc_converged.inputs.parameters.get_dict()) # reuse old settings
# add JIJRAD (remember: in alat units)
Jij_params.set_value('JIJRAD', 1.5)
# add 'XCPL' runopt to list of runopts
runopts = Jij_params.get_value('RUNOPT')
runopts.append('XCPL ')
Jij_params.set_value('RUNOPT', runopts)
# now use updated parameters
builder.parameters = Dict(dict=qdos_params.get_dict())
The calculation is then ready to be submitted:
# submit calculation
kkrcalc = submit(builder)
The result of the calculation will then contain the Jijatom.* files in the
retrieved node and the shells.dat files which allows to map the values of the
exchange interaction to equivalent positions in the different shells.
KKR impurity calculation
Plugin: kkr.kkrimp
- Four input nodes:
parameters, optional: KKR parameter fitting the requirements for a KKRimp calculation (Dict)Only one of
impurity_potential: starting potential for the impurity run (SingleFileData)parent_folder: previous KKRimp parent calulation folder (RemoteFolder)
code: KKRimp code node (code)host_Greenfunction_folder: KKR parent calulation folder containing the writeout of the host’s Green function files (RemoteFolder)
Note
If no parameters node is given then the default values are extracted from the host_Greenfunction calculation.
- Three output nodes:
remote_folder(RemoteData)retrieved(FolderData)output_parameters(Dict)
Note
The parent calculation can be one of the following:
Voronoi calculation, initial calculation starting from structure
previous KKR calculation, e.g. preconverged calculation
The necessary structure information is always extracted from the voronoi parent calculation. In case of a continued calculation the voronoi parent is recuresively searched for.
Create impurity potential
Now the starting potential for the impurity calculation needs to be generated. This means that we need to create an auxiliary structure which contians the impurity in the system where we want to embed it. Then we run a Voronoi calculation to create the starting potential. Here we use the example of a Au impurity embedded into bulk Cu.
The impurity code expects an aiida SingleFileData object that contains the impurity
potential. This is finally constructed using the neworder_potential_wf workfunction
from aiida_kkr.tools.common_workfunctions.
We start with the creation of the auxiliary styructure:
# use an aiida calcfunction to keep track of the provenance
from aiida.engine import calcfunction
@calcfunction
def change_struc_imp_aux_wf(struc, imp_info): # Note: works for single imp at center only!
from aiida.common.constants import elements as PeriodicTableElements
_atomic_numbers = {data['symbol']: num for num, data in PeriodicTableElements.iteritems()}
new_struc = StructureData(cell=struc.cell)
isite = 0
for site in struc.sites:
sname = site.kind_name
kind = struc.get_kind(sname)
pos = site.position
zatom = _atomic_numbers[kind.get_symbols_string()]
if isite == imp_info.get_dict().get('ilayer_center'):
zatom = imp_info.get_dict().get('Zimp')[0]
symbol = PeriodicTableElements.get(zatom).get('symbol')
new_struc.append_atom(position=pos, symbols=symbol)
isite += 1
return new_struc
new_struc = change_struc_imp_aux_wf(voro_calc.inputs.structure, imp_info)
Note
This functionality is alreadyincorporated in the kkr_imp_wc workflow.
Then we run the Voronoi calculation for auxiliary structure to create the impurity starting potential:
codename = 'voronoi@localhost'
code = Code.get_from_string(codename)
builder = code.get_builder()
builder.metadata.options = {'resources': {'num_machines':1, 'tot_num_mpiprocs':1}}
builder.structure = new_struc
builder.parameters = kkrcalc_converged.inputs.parameters
voro_calc_aux = submit(builder)
Now we create the impurity starting potential using the converged host potential for the surrounding of the impurity and the new Au impurity startpot:
from aiida_kkr.tools.common_workfunctions import neworder_potential_wf
potname_converged = kkrcalc_converged._POTENTIAL
potname_imp = 'potential_imp'
neworder_pot1 = [int(i) for i in loadtxt(GF_host_calc.outputs.retrieved.get_abs_path('scoef'), skiprows=1)[:,3]-1]
potname_impvorostart = voro_calc_aux._OUT_POTENTIAL_voronoi
replacelist_pot2 = [[0,0]]
settings_dict = {'pot1': potname_converged, 'out_pot': potname_imp, 'neworder': neworder_pot1,
'pot2': potname_impvorostart, 'replace_newpos': replacelist_pot2, 'label': 'startpot_KKRimp',
'description': 'starting potential for Au impurity in bulk Cu'}
settings = Dict(dict=settings_dict)
startpot_Au_imp_sfd = neworder_potential_wf(settings_node=settings,
parent_calc_folder=kkrcalc_converged.outputs.remote_folder,
parent_calc_folder2=voro_calc_aux.outputs.remote_folder)
Create and submit initial KKRimp calculation
Now we create a new impurity calculation, set all input nodes and submit the calculation
to preconverge the impurity potential (Au embedded into Cu ulk host as described in the
impurity_info node):
# needed to link to host GF writeout calculation
GF_host_output_folder = GF_host_calc.outputs.remote_folder
# create new KKRimp calculation
from aiida_kkr.calculations.kkrimp import KkrimpCalculation
kkrimp_calc = KkrimpCalculation()
builder = Code.get_from_string('KKRimp@my_mac')
builder.code(kkrimp_code)
builder.host_Greenfunction_folder = GF_host_output_folder
builder.impurity_potential = startpot_Au_imp_sfd
builder.resources = resources
# first set 20 simple mixing steps
kkrimp_params = kkrparams(params_type='kkrimp')
kkrimp_params.set_multiple_values(SCFSTEPS=20, IMIX=0, MIXFAC=0.05)
ParamsKKRimp = Dict(dict=kkrimp_params.get_dict())
bilder.parameters = ParamsKKRimp
# submit calculation
kkrimp_calc = submit(builder)
Restart KKRimp calculation from KKRimp parent
Here we demonstrate how to restart a KKRimp calculation from a parent calculation from which the starting potential is extracted autimatically. This is used to compute the converged impurity potential starting from the previous preconvergence step:
builder = kkrimp_code.get_builder()
builder.parent_calc_folder = kkrimp_calc.outputs.remote_folder
builder.metadata.options = {'resources': resources}
builder.host_Greenfunction_folder = kkrimp_calc.inputs.GFhost_folder
kkrimp_params = kkrparams(params_type='kkrimp', **kkrimp_calc.inputs.parameters.get_dict())
kkrimp_params.set_multiple_values(SCFSTEPS=99, IMIX=5, MIXFAC=0.05)
ParamsKKRimp = Dict(dict=kkrimp_params.get_dict())
builder.parameters = ParamsKKRimp
# submit
kkrimp_calc_converge = submit(builder)
Impurity DOS
create final imp DOS (new host GF for DOS contour, then KKRimp calc using converged potential)
first prepare host GF with DOS contour:
params = kkrparams(**GF_host_calc.inputs.parameters.get_dict())
params.set_multiple_values(EMIN=-0.2, EMAX=GF_host_calc.res.fermi_energy+0.1, NPOL=0, NPT1=0, NPT2=101, NPT3=0)
ParaNode = Dict(dict=params.get_dict())
code = GF_host_calc.inputs.code # take the same code as in the calculation before
builder= code.new_calc()
resources = GF_host_calc.get_resources()
builder.resources = resources
builder.parameters = ParaNode
builder.parent_folder = kkr_converged_parent_folder
builder.impurity_info = GF_host_calc.inputs.impurity_info
GF_host_doscalc = submit(builder)
Then we run the KKRimp step using the converged potential (via the parent_calc_folder
node) and the host GF which contains the DOS contour information (via host_Greenfunction_folder):
builder = kkrimp_calc_converge.inputs.code.get_builder()
builder.host_Greenfunction_folder(GF_host_doscalc.outputs.remote_folder)
builder.parent_calc_folder(kkrimp_calc_converge.outputs.remote_folder)
builder.resources(kkrimp_calc_converge.get_resources())
params = kkrparams(params_type='kkrimp', **kkrimp_calc_converge.inputs.parameters.get_dict())
params.set_multiple_values(RUNFLAG=['lmdos'], SCFSTEPS=1)
ParaNode = Dict(dict=params.get_dict())
builder.parameters(ParaNode)
kkrimp_doscalc = submit(builder)
Finally we plot the DOS:
# get interpolated DOS from GF_host_doscalc calculation:
from masci_tools.io.common_functions import interpolate_dos
dospath_host = GF_host_doscalc.outputs.retrieved.get_abs_path('')
ef, dos, dos_interpol = interpolate_dos(dospath_host, return_original=True)
dos, dos_interpol = dos[0], dos_interpol[0]
# read in impurity DOS
from numpy import loadtxt
impdos0 = loadtxt(kkrimp_doscalc.outputs.retrieved.get_abs_path('out_lmdos.interpol.atom=01_spin1.dat'))
impdos1 = loadtxt(kkrimp_doscalc.outputs.retrieved.get_abs_path('out_lmdos.interpol.atom=13_spin1.dat'))
# sum over spins:
impdos0[:,1:] = impdos0[:,1:]*2
impdos1[:,1:] = impdos1[:,1:]*2
# plot bulk and impurity DOS
from matplotlib.pyplot import figure, fill_between, plot, legend, title, axhline, axvline, xlim, ylim, ylabel, xlabel, title, show
figure()
fill_between((dos_interpol[:,0]-ef)*13.6, dos_interpol[:,1]/13.6, color='lightgrey', lw=0, label='bulk Cu')
plot((impdos0[:,0]-ef)*13.6, impdos0[:,1]/13.6, label='Au imp')
plot((impdos0[:,0]-ef)*13.6, impdos1[:,1]/13.6, label='1st Cu neighbor')
plot((impdos0[:,0]-ef)*13.6, (impdos1[:,1]-dos_interpol[:,1])/dos_interpol[:,1], '--', label='relative difference in 1st Cu neighbor')
legend()
title('DOS of Au impurity embedded into bulk Cu')
axhline(0, lw=1, color='grey')
axvline(0, lw=1, color='grey')
xlim(-8, 1)
ylim(-0.5,8.5)
xlabel('E-E_F (eV)')
ylabel('DOS (states/eV)')
show()
Which should look like this:
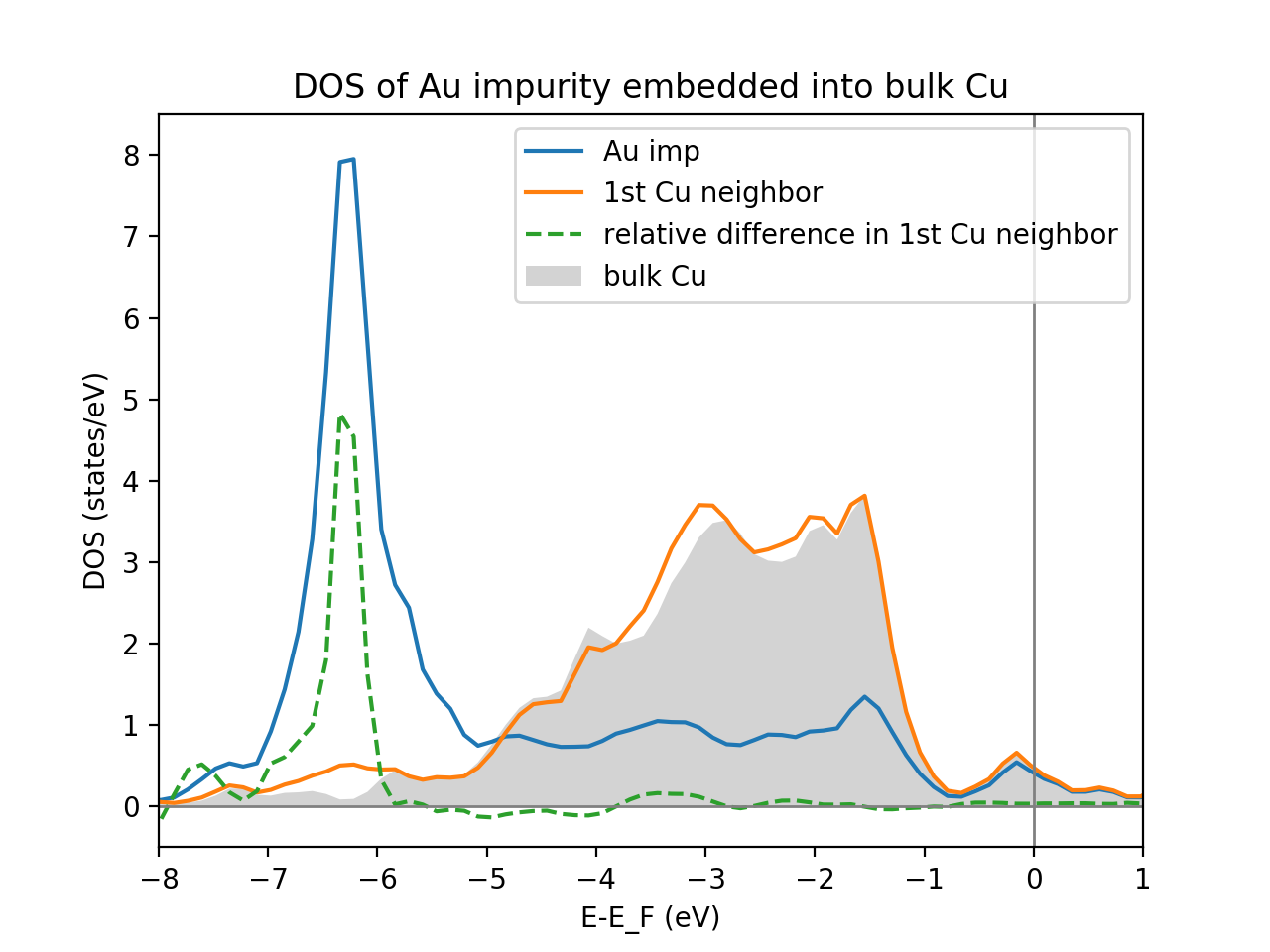
KKR calculation importer
Only functional in version below 1.0
Plugin kkr.kkrimporter
The calculation importer can be used to import a already finished KKR calculation to the aiida dbatabase. The KKRimporterCalculation takes the inputs
code: KKR code installation on the computer from which the calculation is imported
computer: computer on which the calulation has been performed
resources: resources used in the calculation
remote_workdir: remote abolute path oncomputerto the path where the calculation has been performed
input_file_names: dictionary of input file names
output_file_names, optional: dictionary of output file names
and mimicks a KKR calculation (i.e. stores KKR parameter set in node parameters and
the extracted aiida StructureData node structure as inputs and creates
remote_folder, retrieved and output_parameters output nodes).
A KKRimporter calculation can then be used like a KKR claculation to continue
calculations with correct provenance tracking in the database.
Note
At least
input_fileandpotential_fileneed to be given ininput_file_names.Works also if output was a Jij calculation, then
Jijatom.*andshells.datfiles are retreived as well.
Example on how to use the calculation importer:
# Load the KKRimporter class
from aiida.orm import CalculationFactory
KkrImporter = CalculationFactory('kkr.kkrimporter')
# Load the Code node representative of the one used to perform the calculations
from aiida.orm.code import Code
code = Code.get_from_string('KKRcode@my_mac')
# Get the Computer node representative of the one the calculations were run on
computer = code.get_remote_computer()
# Define the computation resources used for the calculations
resources = {'num_machines': 1, 'num_mpiprocs_per_machine': 1}
# Create calculation
calc1 = KkrImporter(computer=computer,
resources=resources,
remote_workdir='<absolute-remote-path-to-calculation>',
input_file_names={'input_file':'inputcard', 'potential_file':'potential', 'shapefun_file':'shapefun'},
output_file_names={'out_potential_file':'potential'})
# Link the code that was used to run the calculations.
calc1.use_code(code)
# Get the computer's transport and create an instance.
from aiida.backends.utils import get_authinfo, get_automatic_user
authinfo = get_authinfo(computer=computer, aiidauser=get_automatic_user())
transport = authinfo.get_transport()
# Open the transport for the duration of the immigrations, so it's not
# reopened for each one. This is best performed using the transport's
# context guard through the ``with`` statement.
with transport as open_transport:
# Parse the calculations' input files to automatically generate and link the
# calculations' input nodes.
calc1.create_input_nodes(open_transport)
# Store the calculations and their input nodes and tell the daeomon the output
# is ready to be retrieved and parsed.
calc1.prepare_for_retrieval_and_parsing(open_transport)
After the calculation has finished the following nodes should appear in the aiida database:
$ verdi calculation show <pk-to-imported-calculation>
----------- ------------------------------------
type KkrImporterCalculation
pk 22121
uuid 848c2185-8c82-44cd-ab67-213c20aaa414
label
description
ctime 2018-04-24 15:29:42.136154+00:00
mtime 2018-04-24 15:29:48.496421+00:00
computer [1] my_mac
code KKRcode
----------- ------------------------------------
##### INPUTS:
Link label PK Type
------------ ----- -------------
parameters 22120 Dict
structure 22119 StructureData
##### OUTPUTS:
Link label PK Type
----------------- ----- -------------
remote_folder 22122 RemoteData
retrieved 22123 FolderData
output_parameters 22124 Dict
##### LOGS:
There are 1 log messages for this calculation
Run 'verdi calculation logshow 22121' to see them
Example scripts
Here is a small collection of example scripts.
Scripts need to be updated for new version (>1.0)
Full example Voronoi-KKR-KKRimp
Compact script starting with structure setup, then voronoi calculation, followed by initial KKR claculation which is then continued for convergence. The converged calculation is then used to write out the host GF and a simple inmpurity calculation is performed.
Download: this example script
#!/usr/bin/env python
# connect to aiida db
from aiida import load_profile
load_profile()
# load essential aiida classes
from aiida.orm import Code
from aiida.orm import DataFactory
StructureData = DataFactory('structure')
Dict = DataFactory('parameter')
# load kkrparms class which is a useful tool to create the set of input parameters for KKR-family of calculations
from aiida_kkr.tools.kkr_params import kkrparams
# load some python modules
from numpy import array
# helper function
def wait_for_it(calc, maxwait=300):
from time import sleep
N = 0
print 'start waiting for calculation to finish'
while not calc.has_finished() and N<(maxwait/2.):
N += 1
if N%5==0:
print('.')
sleep(2.)
print('waiting done after {} seconds: {} {}'.format(N*2, calc.has_finished(), calc.has_finished_ok()))
###################################################
# initial structure
###################################################
# create Copper bulk aiida Structure
alat = 3.61 # lattice constant in Angstroem
bravais = alat*array([[0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5]]) # Bravais matrix in Ang. units
Cu = StructureData(cell=bravais)
Cu.append_atom(position=[0,0,0], symbols='Cu')
###################################################
# Voronoi step (preparation of starting potential)
###################################################
# create empty set of KKR parameters (LMAX cutoff etc. ) for voronoi code
params = kkrparams(params_type='voronoi')
# and set at least the mandatory parameters
params.set_multiple_values(LMAX=2, NSPIN=1, RCLUSTZ=2.3)
# finally create an aiida Dict node and fill with the dictionary of parameters
ParaNode = Dict(dict=params.get_dict())
# choose a valid installation of the voronoi code
### !!! adapt to your code name !!! ###
codename = 'voronoi@my_mac'
code = Code.get_from_string(codename)
# create new instance of a VoronoiCalculation
voro_calc = code.new_calc()
# and set resources that will be used (here serial job)
voro_calc.set_resources({'num_machines':1, 'tot_num_mpiprocs':1})
### !!! use queue name if necessary !!! ###
# voro_calc.set_queue_name('<quene_name>')
# then set structure and input parameter
voro_calc.use_structure(Cu)
voro_calc.use_parameters(ParaNode)
# store all nodes and submit the calculation
voro_calc.store_all()
voro_calc.submit()
wait_for_it(voro_calc)
# for future reference
voronoi_calc_folder = voro_calc.outputs.remote_folder
voro_params = voro_calc.inputs.parameters
###################################################
# KKR step (20 iterations simple mixing)
###################################################
# create new set of parameters for a KKR calculation and fill with values from previous voronoin calculation
params = kkrparams(params_type='kkr', **voro_params.get_dict())
# and set the missing values
params.set_multiple_values(RMAX=7., GMAX=65.)
# choose 20 simple mixing iterations first to preconverge potential (here 5% simple mixing)
params.set_multiple_values(NSTEPS=20, IMIX=0, STRMIX=0.05)
# create aiida Dict node from the KKR parameters
ParaNode = Dict(dict=params.get_dict())
# get KKR code and create new calculation instance
### !!! use your code name !!! ###
code = Code.get_from_string('KKRcode@my_mac')
kkr_calc = code.new_calc()
# set input Parameter, parent calulation (previous voronoi calculation), computer resources
kkr_calc.use_parameters(ParaNode)
kkr_calc.use_parent_folder(voronoi_calc_folder)
kkr_calc.set_resources({'num_machines': 1, 'num_mpiprocs_per_machine':1})
### !!! use queue name if necessary !!! ###
# kkr_calc.set_queue_name('<quene_name>')
# store nodes and submit calculation
kkr_calc.store_all()
kkr_calc.submit()
# wait for calculation to finish
wait_for_it(kkr_calc)
###################################################
# 2nd KKR step (continued from previous KKR calc)
###################################################
# create new KKR calculation instance to continue KKR ontop of a previous KKR calclation
kkr_calc_continued = code.new_calc()
# reuse old KKR parameters and update scf settings (default is NSTEPS=1, IMIX=0)
params.set_multiple_values(NSTEPS=50, IMIX=5)
# and create aiida Dict node
ParaNode = Dict(dict=params.get_dict())
# then set input nodes for calculation
kkr_calc_continued.use_code(code)
kkr_calc_continued.use_parameters(ParaNode)
kkr_calc_parent_folder = kkr_calc.outputs.remote_folder # parent remote folder of previous calculation
kkr_calc_continued.use_parent_folder(kkr_calc_parent_folder)
kkr_calc_continued.set_resources({'num_machines': 1, 'num_mpiprocs_per_machine':1})
### !!! use queue name if necessary !!! ###
# kkr_calc_continued.set_queue_name('<quene_name>')
# store input nodes and submit calculation
kkr_calc_continued.store_all()
kkr_calc_continued.submit()
# wait for calculation to finish
wait_for_it(kkr_calc_continued)
###################################################
# writeout host GF (using converged calculation)
###################################################
# take remote folder of converged calculation to reuse setting and write out Green function and tmat of the crystalline host system
kkr_converged_parent_folder = kkr_calc_continued.outputs.remote_folder
# extreact kkr calculation from parent calculation folder
kkrcalc_converged = kkr_converged_parent_folder.get_inputs()[0]
# extract parameters from parent calculation and update RUNOPT for KKRFLEX option
kkr_params_dict = kkrcalc_converged.inputs.parameters.get_dict()
kkr_params_dict['RUNOPT'] = ['KKRFLEX']
# create aiida Dict node with set parameters that are updated compared to converged parent kkr calculation
ParaNode = Dict(dict=kkr_params_dict)
# create new KKR calculation
code = kkrcalc_converged.get_code() # take the same code as in the calculation before
GF_host_calc= code.new_calc()
# set resources, Parameter Node and parent calculation
resources = kkrcalc_converged.get_resources()
GF_host_calc.set_resources(resources)
GF_host_calc.use_parameters(ParaNode)
GF_host_calc.use_parent_folder(kkr_converged_parent_folder)
### !!! use queue name if necessary !!! ###
# GF_host_calc.set_queue_name('<quene_name>')
# prepare impurity_info node containing the information about the impurity cluster
imp_info = Dict(dict={'Rcut':1.01, 'ilayer_center':0, 'Zimp':[79.]})
# set impurity info node to calculation
GF_host_calc.use_impurity_info(imp_info)
# store input nodes and submit calculation
GF_host_calc.store_all()
GF_host_calc.submit()
# wait for calculation to finish
wait_for_it(GF_host_calc)
######################################################################
# KKRimp calculation (20 simple mixing iterations for preconvergence)
######################################################################
# first create impurity start pot using auxiliary voronoi calculation
# creation of the auxiliary styructure:
# use an aiida workfunction to keep track of the provenance
from aiida.work import workfunction as wf
@wf
def change_struc_imp_aux_wf(struc, imp_info): # Note: works for single imp at center only!
from aiida.common.constants import elements as PeriodicTableElements
_atomic_numbers = {data['symbol']: num for num, data in PeriodicTableElements.iteritems()}
new_struc = StructureData(cell=struc.cell)
isite = 0
for site in struc.sites:
sname = site.kind_name
kind = struc.get_kind(sname)
pos = site.position
zatom = _atomic_numbers[kind.get_symbols_string()]
if isite == imp_info.get_dict().get('ilayer_center'):
zatom = imp_info.get_dict().get('Zimp')[0]
symbol = PeriodicTableElements.get(zatom).get('symbol')
new_struc.append_atom(position=pos, symbols=symbol)
isite += 1
return new_struc
new_struc = change_struc_imp_aux_wf(voro_calc.inputs.structure, imp_info)
# then Voronoi calculation for auxiliary structure
### !!! use your code name !!! ###
codename = 'voronoi@my_mac'
code = Code.get_from_string(codename)
voro_calc_aux = code.new_calc()
voro_calc_aux.set_resources({'num_machines':1, 'tot_num_mpiprocs':1})
voro_calc_aux.use_structure(new_struc)
voro_calc_aux.use_parameters(kkrcalc_converged.inputs.parameters)
voro_calc_aux.store_all()
voro_calc_aux.submit()
### !!! use queue name if necessary !!! ###
# voro_calc_aux.set_queue_name('<quene_name>')
# wait for calculation to finish
wait_for_it(voro_calc_aux)
# then create impurity startpot using auxiliary voronoi calc and converged host potential
from aiida_kkr.tools.common_workfunctions import neworder_potential_wf
potname_converged = kkrcalc_converged._POTENTIAL
potname_imp = 'potential_imp'
neworder_pot1 = [int(i) for i in loadtxt(GF_host_calc.outputs.retrieved.get_abs_path('scoef'), skiprows=1)[:,3]-1]
potname_impvorostart = voro_calc_aux._OUT_POTENTIAL_voronoi
replacelist_pot2 = [[0,0]]
settings_dict = {'pot1': potname_converged, 'out_pot': potname_imp, 'neworder': neworder_pot1,
'pot2': potname_impvorostart, 'replace_newpos': replacelist_pot2, 'label': 'startpot_KKRimp',
'description': 'starting potential for Au impurity in bulk Cu'}
settings = Dict(dict=settings_dict)
startpot_Au_imp_sfd = neworder_potential_wf(settings_node=settings,
parent_calc_folder=kkrcalc_converged.out.remote_folder,
parent_calc_folder2=voro_calc_aux.out.remote_folder)
# now create KKRimp calculation and run first (some simple mixing steps) calculation
# needed to link to host GF writeout calculation
GF_host_output_folder = GF_host_calc.out.remote_folder
# create new KKRimp calculation
from aiida_kkr.calculations.kkrimp import KkrimpCalculation
kkrimp_calc = KkrimpCalculation()
### !!! use your code name !!! ###
kkrimp_code = Code.get_from_string('KKRimp@my_mac')
kkrimp_calc.use_code(kkrimp_code)
kkrimp_calc.use_host_Greenfunction_folder(GF_host_output_folder)
kkrimp_calc.use_impurity_potential(startpot_Au_imp_sfd)
kkrimp_calc.set_resources(resources)
kkrimp_calc.set_computer(kkrimp_code.get_computer())
# first set 20 simple mixing steps
kkrimp_params = kkrparams(params_type='kkrimp')
kkrimp_params.set_multiple_values(SCFSTEPS=20, IMIX=0, MIXFAC=0.05)
ParamsKKRimp = Dict(dict=kkrimp_params.get_dict())
kkrimp_calc.use_parameters(ParamsKKRimp)
# store and submit
kkrimp_calc.store_all()
kkrimp_calc.submit()
# wait for calculation to finish
wait_for_it(kkrimp_calc)
###################################################
# continued KKRimp calculation until convergence
###################################################
kkrimp_calc_converge = kkrimp_code.new_calc()
kkrimp_calc_converge.use_parent_calc_folder(kkrimp_calc.out.remote_folder)
kkrimp_calc_converge.set_resources(resources)
kkrimp_calc_converge.use_host_Greenfunction_folder(kkrimp_calc.inputs.GFhost_folder)
kkrimp_params = kkrparams(params_type='kkrimp', **kkrimp_calc.inputs.parameters.get_dict())
kkrimp_params.set_multiple_values(SCFSTEPS=99, IMIX=5, MIXFAC=0.05)
ParamsKKRimp = Dict(dict=kkrimp_params.get_dict())
kkrimp_calc_converge.use_parameters(ParamsKKRimp)
### !!! use queue name if necessary !!! ###
# kkrimp_calc_converge.set_queue_name('<quene_name>')
# store and submit
kkrimp_calc_converge.store_all()
kkrimp_calc_converge.submit()
wait_for_it(kkrimp_calc_converge)
KKRimp DOS (starting from converged parent KKRimp calculation)
Script running host GF step for DOS contour first before running KKRimp step and plotting.
Download: this example script
#!/usr/bin/env python
# connect to aiida db
from aiida import load_profile
load_profile()
# load essential aiida classes
from aiida.orm import DataFactory, load_node
Dict = DataFactory('parameter')
# some settings:
#DOS contour (in Ry units), emax=EF+dE_emax:
emin, dE_emax, npt = -0.2, 0.1, 101
# kkrimp parent (converged imp pot, needs to tbe a KKRimp calculation node)
kkrimp_calc_converge = load_node(25025)
# derived quantities:
GF_host_calc = kkrimp_calc_converge.inputs.GFhost_folder.inputs.remote_folder
kkr_converged_parent_folder = GF_host_calc.inputs.parent_calc_folder
# helper function
def wait_for_it(calc, maxwait=300):
from time import sleep
N = 0
print 'start waiting for calculation to finish'
while not calc.has_finished() and N<(maxwait/2.):
N += 1
if N%5==0:
print('.')
sleep(2.)
print('waiting done after {} seconds: {} {}'.format(N*2, calc.has_finished(), calc.has_finished_ok()))
################################################################################################
# first host GF with DOS contour
from aiida_kkr.tools.kkr_params import kkrparams
params = kkrparams(**GF_host_calc.inputs.parameters.get_dict())
params.set_multiple_values(EMIN=emin, EMAX=GF_host_calc.res.fermi_energy+dE_emax, NPOL=0, NPT1=0, NPT2=npt, NPT3=0)
ParaNode = Dict(dict=params.get_dict())
code = GF_host_calc.get_code() # take the same code as in the calculation before
GF_host_doscalc= code.new_calc()
resources = GF_host_calc.get_resources()
GF_host_doscalc.set_resources(resources)
GF_host_doscalc.use_parameters(ParaNode)
GF_host_doscalc.use_parent_folder(kkr_converged_parent_folder)
GF_host_doscalc.use_impurity_info(GF_host_calc.inputs.impurity_info)
# store and submit
GF_host_doscalc.store_all()
GF_host_doscalc.submit()
# wait for calculation to finish
print 'host GF calc for DOS contour'
wait_for_it(GF_host_doscalc)
# then KKRimp step using the converged potential
kkrimp_doscalc = kkrimp_calc_converge.get_code().new_calc()
kkrimp_doscalc.use_host_Greenfunction_folder(GF_host_doscalc.out.remote_folder)
kkrimp_doscalc.use_parent_calc_folder(kkrimp_calc_converge.out.remote_folder)
kkrimp_doscalc.set_resources(kkrimp_calc_converge.get_resources())
# set to DOS settings
params = kkrparams(params_type='kkrimp', **kkrimp_calc_converge.inputs.parameters.get_dict())
params.set_multiple_values(RUNFLAG=['lmdos'], SCFSTEPS=1)
ParaNode = Dict(dict=params.get_dict())
kkrimp_doscalc.use_parameters(ParaNode)
# store and submit calculation
kkrimp_doscalc.store_all()
kkrimp_doscalc.submit()
# wait for calculation to finish
print 'KKRimp calc DOS'
wait_for_it(kkrimp_doscalc)
# Finally plot the DOS:
# get interpolated DOS from GF_host_doscalc calculation:
from masci_tools.io.common_functions import interpolate_dos
dospath_host = GF_host_doscalc.out.retrieved.get_abs_path('')
ef, dos, dos_interpol = interpolate_dos(dospath_host, return_original=True)
dos, dos_interpol = dos[0], dos_interpol[0]
# read in impurity DOS
from numpy import loadtxt
impdos0 = loadtxt(kkrimp_doscalc.out.retrieved.get_abs_path('out_lmdos.interpol.atom=01_spin1.dat'))
impdos1 = loadtxt(kkrimp_doscalc.out.retrieved.get_abs_path('out_lmdos.interpol.atom=13_spin1.dat'))
# sum over spins:
impdos0[:,1:] = impdos0[:,1:]*2
impdos1[:,1:] = impdos1[:,1:]*2
# plot bulk and impurity DOS
from matplotlib.pyplot import figure, fill_between, plot, legend, title, axhline, axvline, xlim, ylim, ylabel, xlabel, title, show
figure()
fill_between((dos_interpol[:,0]-ef)*13.6, dos_interpol[:,1]/13.6, color='lightgrey', lw=0, label='bulk Cu')
plot((impdos0[:,0]-ef)*13.6, impdos0[:,1]/13.6, label='Au imp')
plot((impdos0[:,0]-ef)*13.6, impdos1[:,1]/13.6, label='1st Cu neighbor')
plot((impdos0[:,0]-ef)*13.6, (impdos1[:,1]-dos_interpol[:,1])/dos_interpol[:,1], '--', label='relative difference in 1st Cu neighbor')
legend()
title('DOS of Au impurity embedded into bulk Cu')
axhline(0, lw=1, color='grey')
axvline(0, lw=1, color='grey')
xlim(-8, 1)
ylim(-0.5,8.5)
xlabel('E-E_F (eV)')
ylabel('DOS (states/eV)')
show()
KKR bandstructure
Script running a bandstructure calculation for which first from the structure node
the kpoints of the high-symmetry lines are extracted and afterwards the bandstructure
(i.e. qdos) calculation is started. Finally the results are plotted together with
the DOS data (taken from KKRimp DOS preparation step).
Download: this example script
#!/usr/bin/env python
# connect to aiida db
from aiida import load_profile
load_profile()
# load essential aiida classes
from aiida.orm import Code, DataFactory, load_node
StructureData = DataFactory('structure')
Dict = DataFactory('parameter')
# helper function:
def wait_for_it(calc, maxwait=300):
from time import sleep
N = 0
print 'start waiting for calculation to finish'
while not calc.has_finished() and N<(maxwait/2.):
N += 1
if N%5==0:
print('.')
sleep(2.)
print('waiting done after {} seconds: {} {}'.format(N*2, calc.has_finished(), calc.has_finished_ok()))
# some settings (parent calculations):
# converged KKR calculation (taken form bulk Cu KKR example)
kkr_calc_converged = load_node(24951)
# previous DOS calculation started from converged KKR calc (taken from KKRimp DOS example, i.e. GF host calculation with DOS contour)
host_dos_calc = load_node(25030)
# generate kpoints for bandstructure calculation
from aiida_kkr.calculations.voro import VoronoiCalculation
struc, voro_parent = VoronoiCalculation.find_parent_structure(kkr_calc_converged.out.remote_folder)
from aiida.tools.data.array.kpoints import get_explicit_kpoints_path
kpts = get_explicit_kpoints_path(struc).get('explicit_kpoints')
# run bandstructure calculation
# create bandstructure calculation reusing old settings (including same computer and resources in this example)
kkrcode = kkr_calc_converged.get_code()
kkrcalc = kkrcode.new_calc()
kkrcalc.use_kpoints(kpts) # pass kpoints as input
kkrcalc.use_parent_folder(kkr_calc_converged.out.remote_folder)
kkrcalc.set_resources(kkr_calc_converged.get_resources())
# change parameters to qdos settings (E range and number of points)
from aiida_kkr.tools.kkr_params import kkrparams
qdos_params = kkrparams(**kkr_calc_converged.inputs.parameters.get_dict()) # reuse old settings
# reuse the same emin/emax settings as in DOS run (extracted from input parameter node)
qdos_params.set_multiple_values(EMIN=host_dos_calc.inputs.parameters.get_dict().get('EMIN'),
EMAX=host_dos_calc.inputs.parameters.get_dict().get('EMAX'),
NPT2=100)
kkrcalc.use_parameters(Dict(dict=qdos_params.get_dict()))
# store and submit calculation
kkrcalc.store_all()
kkrcalc.submit()
wait_for_it(kkrcalc, maxwait=600)
# plot results
# extract kpoint labels
klbl = kpts.labels
# fix overlapping labels (nicer plotting)
tmp = klbl[2]
tmp = (tmp[0], '\n'+tmp[1]+' ')
klbl[2] = tmp
tmp = klbl[3]
tmp = (tmp[0], ' '+tmp[1])
klbl[3] = tmp
#plotting of bandstructure and previously calculated DOS data
# load DOS data
from masci_tools.io.common_functions import interpolate_dos
dospath_host = host_dos_calc.out.retrieved.get_abs_path('')
ef, dos, dos_interpol = interpolate_dos(dospath_host, return_original=True)
dos, dos_interpol = dos[0], dos_interpol[0]
# load qdos file and reshape
from numpy import loadtxt, sum, log
qdos_file = kkrcalc.out.retrieved.get_abs_path('qdos.01.1.dat')
q = loadtxt(qdos_file)
nepts = len(set(q[:,0]))
data = q[:,5:].reshape(nepts, len(q)/nepts, -1)
e = (q[::len(q)/nepts, 0]-ef)*13.6
# plot bandstructure
from matplotlib.pyplot import figure, pcolormesh, show, xticks, ylabel, axhline, axvline, gca, title, plot, ylim, xlabel, suptitle
figure(figsize=((8, 4.8)))
pcolormesh(range(len(q)/nepts), e, log(sum(abs(data), axis=2)), lw=0)
xticks([i[0] for i in klbl], [i[1] for i in klbl])
ylabel('E-E_F (eV)')
axhline(0, color='lightgrey', lw=1)
title('band structure')
# plot DOS on right hand side of bandstructure plot
axBand = gca()
from mpl_toolkits.axes_grid1 import make_axes_locatable
divider = make_axes_locatable(axBand)
axDOS = divider.append_axes("right", 1.2, pad=0.1, sharey=axBand)
plot(dos_interpol[:,1]/13.6, (dos_interpol[:,0]-ef)*13.6)
ylim(e.min(), e.max())
axhline(0, color='grey', lw=1)
axvline(0, color='grey', lw=1)
axDOS.yaxis.set_tick_params(labelleft=False, labelright=True, right=True, left=False)
xlabel('states/eV')
title('DOS')
suptitle(struc.get_formula(), fontsize=16)
show()